|
|
|
|
|
Galleria Tassonomica
di
Natura Mediterraneo
|
|
|
| Autore |
 Discussione Discussione  |
|
|
ang
Moderatore


Città: roma
Regione: Lazio

11361 Messaggi
Tutti i Forum |
 Inserito il - 17 giugno 2013 : 22:13:52 Inserito il - 17 giugno 2013 : 22:13:52


|
ciao a tutti
vi segnalo un lavoro molto interessante di jan sauer e bernhard hausdorf, a comparison of dna-based methods for delimiting species in a cretan land snail radiation reveals shortcomings of exclusively molecular taxonomy, cladistics 28(3) 300-316. provo a riassumerlo sperando di non travisarne il messaggio. gli autori mettono a confronto diversi approcci e criteri utilizzati per discriminare entità a livello specifico tramite le tecniche molecolari, utilizzando il genere Xerocrassa nell'isola di creta come banco di prova. i metodi e i criteri vengono giudicati in base alla loro capacità di identificare correttamente esemplari classificati con metodi convenzionali, ovvero tramite la morfologia della conchiglia e dell'anatomia dei genitali. non posso entrare nel merito delle varie tecniche perché non ne ho le competenze, comunque leggendo la discussione il quadro che emerge è piuttosto desolante. la maggior parte degli approcci ha una percentuale di esemplari classificati correttamente a di sotto del 50%. in alcuni casi si arriva quasi all'80%, ma ci sono comunque diversi errori di lumping/splitting e di esempalri allocati in cluster errati. il criterio meno affidabile risulta essere quello della soglia fissa di differenza tra due sequenze genetiche; spesso questa soglia vine fissata tra il 3 e il 5%, ma quello che emerge da questo studio è che non esiste un "numero magico" universalmente valido che possa costituire una soglia oltre la quale si può parlare di specie distinte. questo risultato mi sembra molto ragionevole e credo che un po' tutti avessimo questa sensazione. un fatto importante sottolineato dagli autori è che secondo loro le tecniche molecolari potrebbero essere più efficaci se si potessero prendere in considerazione i geni coinvolti nei processi di speciazione che sono quasi sempre ignoti, tra cui quelli che influenzano l'apparato riproduttivo o i rituali di corteggiamento/accoppiamento. secondo gli autori è questo il motivo della ancora persistente superiorità dei metodi morfologici convenzionali rispetto alle tecniche molecolari, dato che quello che avviene con il metodo di indagine morfologica è che si vanno a ricercare proprio le differenze a livello riproduttivo. il risultato di questo studio evidenzia uno scetticismo degli autori nei confronti del cosiddetto dna barcoding e considerano le tecniche molecolari come un supporto all'indagine morfologica e non come la soluzione dei problemi sistematici
la conclusione mi sembra condivisibile, anche se in tutto questo c'è un punto debole che ne costituisce la premessa, quello cioè di considerare valide le specie determinate per via morfologica
|
ciao
ang
Anche se nessuno di noi sa esattamente dove sta andando o dove andrà a finire.....comunque lascerà la sua indelebile striscia di bava (Beppe/papuina) |
|
|
PaoloMarenzi
Utente Senior


Città: Cremona
Prov.: Cremona
Regione: Lombardia
2807 Messaggi
Flora e Fauna |
Inserito il - 17 giugno 2013 : 22:22:40
|
| grazie mille per aver riassunto ciò che altrimenti non avrei mai potuto maneggiare...dato che si tratta di una vera patata bollente.... |
amatoeridano.blogspot.it |
 |
|
|
Cmb
Moderatore

Città: Buers
Prov.: Estero
Regione: Austria
12844 Messaggi
Flora e Fauna |
Inserito il - 18 giugno 2013 : 07:43:49
|
In 20xx Hausdorf ha presentato il suo lavoro (non ancora pubblicato) al Museo a Stoccarda - mi sembra il titolo era "Xerocrassa su Creta - 8 o 100 specie".
Arriviamo sempre al problema: "Cos'è una specie" - io posso parlare solo per il genere Limax - e qui si deve guardare la morfologia, l'anatomia, il modo dell'accopiamento e al fine si puo controllare tutto con DNA....
è uscito il nostro giornale della EVMG (No 20  ) - e P. L. Reischütz scrive nel prologo per questa tema... ) - e P. L. Reischütz scrive nel prologo per questa tema...
|
"Good people don't go into government" (D. Trump)

Link - nothing is more dangerous than the truth - solo chi conosce il passato, può capire il presente! - nothing is more dangerous than the truth - solo chi conosce il passato, può capire il presente!
|
|
|
|
fern
Utente Senior
Città: Vicenza
2381 Messaggi
Flora e Fauna |
Inserito il - 18 giugno 2013 : 23:40:58
|
Grazie Ang per averci segnalato questo articolo e questa rivista che stranamente non avevo messo fra quelle "da consultare". Lo leggerò con grande interesse; nel frattempo ricordo che in questa discussione, relativa proprio ad un lavoro di Hausdorf, avevo già sollevato il problema citando altri articoli, per dire che la problematica è tutt'altro che nuova. Personalmente, da quegli articoli e da altri mi sono fatto l'idea che il barcoding rappresenta una semplificazione estrema, che in alcuni casi funziona pure ma non è certo la norma. Questo non mi scandalizza affatto, perché il vero vantaggio della sequenziazione genica consiste per me nella ricostruzione di alberi filogenetici, ovvero dei rapporti di parentela fra diverse popolazioni e taxa, lasciando poi alla "discrezione" dello studioso decidere quali siano specie, quali sottospecie o generi ecc. distinti, integrando a questo scopo tutte le altre informazioni disponibili. Il vero problema è che anche gli alberi filogenetici a volte sono sbagliati, forse più spesso di quanto pensiamo! Ciao,
fern
|
Il n'y a de petit dans la Nature que les petits esprits. |
|
|
|
ang
Moderatore
Città: roma
Regione: Lazio
11361 Messaggi
Tutti i Forum |
Inserito il - 20 giugno 2013 : 13:27:56
|
| Messaggio originario di fern:
mi sono fatto l'idea che il barcoding rappresenta una semplificazione estrema, che in alcuni casi funziona pure ma non è certo la norma
|
a quanto pare sì, ad es. c'è un recente articolo di barco et al sul genere Gibbula nel mediterraneo (v. qui) in cui il barcoding sembra funzionare bene con una soglia del 2-3% sulle sequenze COI |
ciao
ang
Anche se nessuno di noi sa esattamente dove sta andando o dove andrà a finire.....comunque lascerà la sua indelebile striscia di bava (Beppe/papuina) |
|
|
|
fern
Utente Senior
Città: Vicenza
2381 Messaggi
Flora e Fauna |
Inserito il - 04 ottobre 2013 : 00:50:48
|
Avevo promesso una risposta ma mi sono arenato ed ho finito per dimenticarmene. Penso che per rispondere adeguatamente dovrei prendere in considerazione anche i due seguenti lavori:
Reconstructing the evolutionary history of the radiation of the land snail genus Xerocrassa on Crete based on mitochondrial sequences and AFLP markers, BMC Evolutionary Biology 2010, 10:299 (di cui si è già discusso nel forum) e
Sexual selection is involved in speciation in a land snail radiation on Crete, Evolution 63-10 (2009): 2535–2546.
Non l'ho ancora fatto e mi limito ad alcune considerazioni sull'articolo in questione.
Vi si sottolinea che le chiocciole presentano problemi particolari, quali
rapida divergenza nel DNA mitocondriale che produce grandi distanze genetiche sia inter che intra-specifiche e che
tendenza a conservare il polimorfismo ancestrale, per la suddivisione in molte popolazioni isolate.
Questo problema è noto come "incomplete lineage sorting" (non conosco traduzioni italiane, si può dire "campionamento genico incompleto"?) il cui senso è il seguente:
- in ogni popolazione finita finisce per restare un solo allele di un dato gene, almeno per variazioni neutre.
- in popolazioni isolate questo allele sarà in genere diverso (in questo senso si conserva del polimorfismo ancestrale) Questo rischio si presenta in particolare quando si confrontano specie affini o popolazioni di una stessa specie.
Inoltre, quando si ha a che fare con specie simili (per non parlare di sottospecie) esiste il problame dell'introgressione: specie simili possono saltuariamente produrre ibridi provocando a volte una vera e propria "invasione genica", soprattutto a livello mitocondriale. Questi fenomeni possono alterare la corrispondenza fra l'albero filogenetico della specie e quello del gene in questione.
Quali che siano i metodi utilizzati nell'articolo per riconoscere le specie (soglia fissa sulla distanza genetica, semplice o sofisticata o parsimonia, utilizzo di 1 o 2 geni) il risultato sostanzialmente non cambia: come già riassunto da Ang, anche nei casi migliori solo il 50% circa di risultati è corretto.
Altri punti.
| | The 122 cox1 sequences of the Cretan Xerocrassa species were grouped into 51 sets, for which each sequence in a set had at least one other sequence within a threshold distance of 3%. Twenty-seven of these sets included only a single specimen ... |
A parte altre considerazioni, mi sembra qui adombrato un problema di campionamento, non diverso da quello della sistematica tradizionale: casi estremi di un complesso di forme possono sembrare specie distinte: pensiamo a Chilostoma cingulatum gobanzi, inizialmente "species distinctissima", salvo ricredersi davanti alle forme di passaggio che lo uniscono a colubrinum senza soluzione di continuità. Il problema con il DNA mi sembra simile: due popolazioni possono apparire specie distinte solo perché non conosciamo casi intermedi che segnano il passaggio dall'una all'altra, e ciò vale in particolare per le specie distribuite su vasti areali.
| | small peripheral populations, ... may rapidly adapt to specific environmental conditions and evolve into separate species. Nevertheless, they may still be more similar to neighbouring populations of a widespread stem species with regard to many marker loci than these populations are to geographically distant populations of the same species. |
Fig. 1
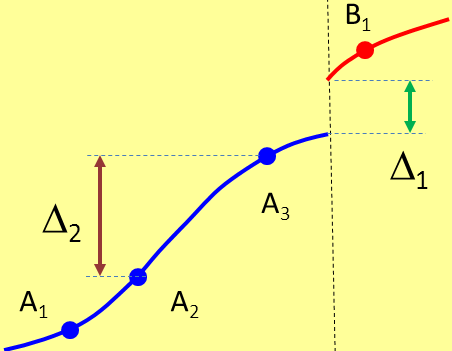
12,49 KB
la linea blu e la linea rossa rappresentano due specie vicarianti: A e B; in orizzontale immaginiamo una "coordinata geografica" e in verticale una misura di distanza genetica. Per separare le due specie basta evidentemente la soglia ∆1, ma se le popolazioni campionate sono quelle rappresentate dai punti, una soglia pari a ∆1 porterebbe a riconoscere 4 specie distinte mentre una soglia di poco inferiore a ∆2 ci porterebbe a concludere che le specie sono sì 2: ma una contenente le popolazioni A1, A2, l'altra con le popolazioni A3, B1.
Purtroppo anche il campionamento più meticoloso può non bastare: che facciamo se una parte della specie A si estingue, come mostra la Fig.2 ?
Fig. 2
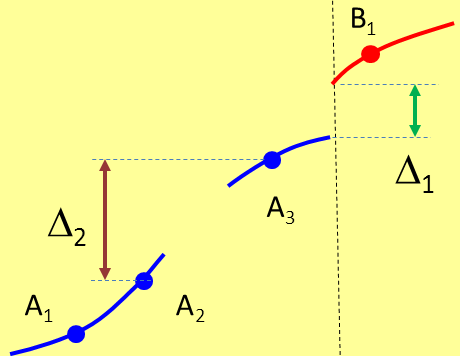
12,26 KB
Ciao,
fern |
|
|
|
fern
Utente Senior
Città: Vicenza
2381 Messaggi
Flora e Fauna |
Inserito il - 08 ottobre 2013 : 19:43:54
|
Vorrei completare il discorso riferendo alcune altre cose che ho reperito sull'argomento, sperando di non tediarvi.
In linea di massima, nei lavori recenti si sparla abbastanza del "barcoding", ovvero della separazione fra specie mediante l'applicazione di una soglia, proprio come sostenuto da Sauer e Hausdorf relativamente alle Xerocrassa di Creta. Sempre in generale si raccomanda di utilizzare metodi basati su alberi filogenetici ed in particolare
• utilizzare più geni ("marker"): si parla di 10, 20 o più!
• utilizzare più metodi (in pratica programmi disponibili sul mercato) che utilizzano diverse assunzioni, per verificare come le diverse ipotesi influenzano il risultato finale. Se i risultati concordano sono "robusti" e ci possiamo fidare, altrimenti (cioè nella maggioranza dei casi) meglio essere cauti.
Insomma, per i teorici di queste cose non esiste un metodo sicuro e si raccomanda la massima prudenza.
Eppure, per alcuni autori ci sarebbe un metodo tanto efficace quanto semplice, basato proprio sull'uso di una soglia: un articolo molto citato è quello di Annette W. Coleman: "Is there a molecular key to the level of "biological species" in eukaryotes? A DNA guide". Mol. Phyl. Evo. 2009.
Per motivi non chiari pare che il gene ITS2 (Internal Transcribed Spacer 2) sia particolarmente appropriato: uno studio del 2007, basato su 1373 specie aveva mostrato un'incredibile correlazione fra le differenze in questo locus e l'appartenenza specie diverse, in particolare certe differenze, dette CBC (Compensatory Base Change) avrebbero un forte valore diagnostico. Si tratta di questo: il gene in questione possiede una struttura secondaria costituita da 4 "eliche" (helix) principali, numerate da I a IV, più alcune secondarie, in cui la singola catena di DNA si avvolge su sè stessa a doppia elica; ciò significa che alcune sequenze di basi sono seguite dalle sequenze complementari (invertite). Una CBC è la sostituzione di una base in un ramo dell'elica, e della sua complementare nell'altro. Nello studio del 2007 si osservò che:
• quando due gruppi differiscono per una CBC, nel 93% dei casi appartengono a specie (tassonomiche) diverse
• in assenza di CBC i due gruppi appartengono alla stessa specie nel 76% dei casi.
Un punto debole di quanto sopra è che la "specie tassonomica" potrebbe non corispondere ad una "specie biologica" e l'articolo della Coleman affronta proprio questo punto utilizzando come "proxy" di interfecondità la compatiblità fra i gameti. Il risultato è notevole: esiste una sequenza di una trentina di basi dell'elica III tale che gli individui che differiscono di almeno 1 CBC sono totalmente incapaci di formare ibridi. Taxa con differenze minori, come una "semi-CBC", mostrano ancora una debole capacità di ibridizzazione. Anche se l'assenza di queste differenze non implica l'appartenenza alla stessa specie, questo fatto mi sembra di straordinaria importanza.
Se ciò è vero, perché in alcuni lavori recenti questi risultati sono ignorati? Non esprimono critiche o riserve, semplicemente lo ignorano oppure è solo un riferimento bibliografico senza nemmeno un commento. Mah!
fern
|
Il n'y a de petit dans la Nature que les petits esprits. |
|
|
|
Cmb
Moderatore
Città: Buers
Prov.: Estero
Regione: Austria
12844 Messaggi
Flora e Fauna |
Inserito il - 08 ottobre 2013 : 20:42:51
|
molto contestato Rupert Sheldrake
Come ho scritto prima: la genetica è un aiuti - ma da solo non puo beatificare....
|
"Good people don't go into government" (D. Trump)
Link - nothing is more dangerous than the truth - solo chi conosce il passato, può capire il presente!
|
|
|
|
ang
Moderatore
Città: roma
Regione: Lazio
11361 Messaggi
Tutti i Forum |
Inserito il - 22 ottobre 2013 : 10:10:06
|
grazie fern per lo schema proposto (fig. 1 e 2) che esemplifica molto bene il rischio che si corre quando si hanno a disposizione pochi campioni
sempre sull'argomento vi segnalo un nuovo interessante lavoro da parte del gruppo di ricercatori coordinato da marco oliverio sullo stato delle ocinebrine del mediterraneo (abstract), in cui i dati molecolari danno un risultato interpretabile in diverse maniere a seconda di dove si pone la soglia; per il momento gli autori non traggono conclusioni definitive ma illustrano due possibili scenari, il primo consistente in 4 cladi, eventualmente suddivisibili in 8 motu (non in senso vaticano ma più propriamente unità tassonomiche rivelate da dati molecolari); nel primo caso si hanno 4 cladi distribuiti in maniera abbastanza sensata da un punto di vista geografico (a parte il clade 3), nel secondo caso si avrebbero 8 presunte specie, al momento però non discriminabili dato che a quanto sembra i dati morfologici non sono determinanti |
ciao
ang
Anche se nessuno di noi sa esattamente dove sta andando o dove andrà a finire.....comunque lascerà la sua indelebile striscia di bava (Beppe/papuina) |
|
|
| |
Discussione |
|
|
|
 Natura Mediterraneo Natura Mediterraneo |
© 2003-2024 Natura Mediterraneo |
|
|
Leps.it | Herp.it | Lynkos.net
|

 Forum
|
Registrati
|
Msg attivi
|
Msg Recenti
|
Msg Pvt
|
Utenti
|
Galleria |
Map |
Forum
|
Registrati
|
Msg attivi
|
Msg Recenti
|
Msg Pvt
|
Utenti
|
Galleria |
Map |
